

Dependence of Absorption and Emission Spectra on Polymorphs of Gold(I) Isocyanide Complexes: Theoretical Study with QM/MM Approach
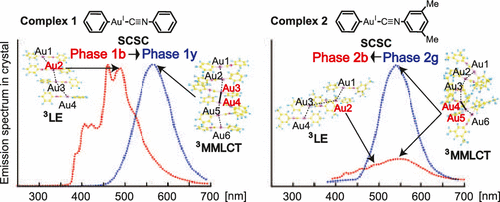
We theoretically investigated phenyl(phenyl isocyanide) gold(I) (PhNC)Au(Ph) 1 and phenyl(dimethylphenyl isocyanide) gold(I) (dimPhNC)Au(Ph) 2 in crystal using our periodic quantum mechanics/molecular mechanics (QM/MM) method based on the self-consistent point charges to elucidate interesting mechano-chemical changes of absorption and emission spectra of 1 and 2 in crystal. To characterize 1 and 2 in crystal, their absorption and emission spectra in crystal were compared to those in gas phase and CHCl3 solvent, where a three-dimensional reference interaction site model self-consistent field (3D-RISM-SCF) was employed to incorporate solvation effect. To investigate the phosphorescence spectrum in crystal, we optimized the geometry of the molecule at the triplet state in crystal which had ground-state geometry because the population of the excited state is generally very small. The QM/MM calculations showed that 1 formed two polymorphs 1b and 1y, 2 formed two polymorphs 2b and 2g, and 1y was more stable than 1b, which agree with the experimental findings. In 1b and 2b, the ligand-to-ligand charge transfer state is the lowest-energy excited state in the absorption, and the π–π* locally excited state on the PhNC moiety is the lowest-energy triplet state in the emission. In 1y and 2g, on the other hand, the metal–metal-to-ligand charge transfer (MMLCT) state is the lowest-energy excited state in both absorption and emission. These characteristic differences between two crystal structures arise from the geometrical features that the Au–Au distance is much shorter in 1y and 2g than in 1b and 1y and the intermolecular torsion angle η between two Au-PhNC moieties is much smaller in 1y and 2g than in 1b and 2b, respectively; the short Au–Au distance raises the energy level of antibonding orbital consisting of two Au dσ orbitals, and the small η angle lowers the energy level of bonding orbital consisting of two PhNC π* orbitals, leading to the presence of a lower-energy MMLCT state. The QM/MM calculations also disclosed intramolecular torsion angle τ between the Ph and PhNC planes and CH−π interaction of the Ph plane significantly influence absorption spectrum. Based on those computational results, discussion is presented on the differences in absorption and emission spectra among gas, solution, and crystal, the assignments of experimentally observed excitation and emission spectra in crystal, and their energy shifts induced by single-crystal-to-single-crystal phase transition.